![]()
![]()
Feature Article
Reporting Unanticipated Problems
to the IRB
(Formerly SAE Reporting)
October 2007 Issue
By Mary Banks, RN, BS, BSN,
IRB Director
Author has nothing to disclose with regards
to commercial support.
Educational Objectives:
At the end of this activity, participants should be able to:
- Define an "unanticipated problem involving risk to subjects or others"
- Differentiate between an adverse event and an unanticipated problem
- Describe the mechanism and form to be used for reporting unanticipated problems to the IRB
- Explain the process for reporting AEs and SAEs as part of the Progress Report
- Introduction
- Regulatory Background and Historical Context
- Unanticipated Problems
- Unanticipated Problems That Are NOT Adverse Events
- Reporting Unanticipated Problems
- Reporting Unanticipated Problems in INSPIR
- Submitting Safety Monitors' Reports
- Progress Reports and Summary Reports
- IRB Reporting to OHRP and the FDA
- Summary
- Quiz
"It would be naive to think that the problems plaguing mankind today can be solved with means and methods which were applied or seemed to work in the past. . " Mikhail Gorbachev, (1988).
 Are
you and your staff overwhelmed with the problem of reporting all individual
serious adverse event (SAEs) to the IRB? Have you been wishing and hoping
for relief from this burden? There is good news for you. The rules have
changed and these changes are expected to result in a significant decrease
in administrative workload for investigators and the IRB at BUMC.
Are
you and your staff overwhelmed with the problem of reporting all individual
serious adverse event (SAEs) to the IRB? Have you been wishing and hoping
for relief from this burden? There is good news for you. The rules have
changed and these changes are expected to result in a significant decrease
in administrative workload for investigators and the IRB at BUMC.
Over the past several years, investigators and IRBs have been inundated with serious adverse event reports (particularly from multi-center trials) that lack detail, context, and information about the relevance of the "event" to the population being studied. Federal regulators have been asked to consider the huge administrative burden required to process these serious adverse event (SAE) reports; as well as the fact that these reports are hindering--rather than helping--IRBs to protect human subjects. Earlier this year, in response to these petitions, both OHRP (Office of Human Research Protections) and the FDA (Food and Drug Administration) issued guidances to clarify their positions with regard to AE (adverse event) reporting. Subsequently, the BUMC IRB has modified its policies for AE and SAE reporting.
Regulatory Background and Historical Context
The January 15, 2007 OHRP “Guidance on Reviewing and Reporting Unanticipated Problems Involving Risks to Subjects or Others and Adverse Events” and the April 2007 FDA [Draft] “Guidance for Clinical Investigators, Sponsors and IRBs, Adverse Event Reporting-Improving Human Subjects Protection” both emphasize that there is NO regulatory requirement for SAEs to be individually reported to IRBs. Instead, they explain that the regulations require that investigators must report to the IRB only those events that represent “any unanticipated problem involving risks to subjects or others”. 45 CFR 46.103(b) (5) and [56.108(b) (1), 312.53(c) (1) (vii), and 312.66].
The term “unanticipated problems involving risks to subjects or others” (henceforth known as “unanticipated problems”) is not defined in either set of regulations. This lack of clarity, combined with complex reporting pathways developed by sponsors and IRBs over the years, have resulted in a standard of practice that exceeds the regulatory requirements, and yet doesn’t offer any benefits in terms of improved surveillance and increased safety. Many IRBs, including the BUMC IRB, have adopted policies that require all SAEs to be individually reported to the IRB. These SAE reports, while excessive in quantity, are usually poor in quality and provide little usable information to the IRB. These guidances make it clear that, while the requirement for reporting all SAEs to local IRBs may represent institutional policy, it is not and has never been, a regulatory requirement.
 The
OHRP and the FDA regulations require that unanticipated problems
be promptly reported to the IRB. So what exactly is an unanticipated
problem? The guidances state that in order for any incident,
experience, or outcome to be an unanticipated problem,
it must meet all three of the following criteria:
The
OHRP and the FDA regulations require that unanticipated problems
be promptly reported to the IRB. So what exactly is an unanticipated
problem? The guidances state that in order for any incident,
experience, or outcome to be an unanticipated problem,
it must meet all three of the following criteria:
- be unexpected (in terms of nature, severity, or frequency)
given (a) the research procedures that are described in the protocol-related
documents (such as the IRB-approved research protocol and informed consent
document); and (b) the characteristics of the subject population being
studied; AND
- be related or possibly related to participation in the research
(in this guidance document, possibly related means there is
a reasonable possibility that the incident, experience, or outcome may
have been caused by the procedures involved in the research); AND
- suggests that the research places subjects or others at a greater risk of harm (including physical, psychological, economic, or social harm) than was previously known or recognized. (Note: OHRP specifies that all incidents, experiences or outcomes that are unexpected, AND related or possibly related AND are SERIOUS automatically meet this third criteria.)
Unanticipated Problems That Are NOT Adverse Events
In its guidance, OHRP has defined an adverse event (AE) as, “any untoward or unfavorable medical occurrence in a human subject, including any abnormal sign (for example, abnormal physical exam or laboratory finding), symptom, or disease, temporally associated with the subject’s participation in the research, whether or not considered related to the subject’s participation in the research.” (This is modified from the definition of adverse events in the 1996 International Conference on Harmonization E-6 Guidelines for Good Clinical Practice.) AEs encompass both physical and psychological harm and can occur in biomedical and social / behavioral research. SAEs are a subgroup of AEs that are serious because they are associated with greater harm to subjects. (Unless otherwise specified, in the remainder of this article, the term AE will be used as an umbrella term that includes all adverse events, both serious and non-serious).
 Occasionally
an incident, experience, or outcome occurs in research that does NOT meet
this definition of an adverse event. Consider, for example, if Charlie,
a study coordinator, leaves his laptop containing subjects’ identities
and social security numbers on the MBTA. This event would not meet the
definition of an adverse event, but would be a reportable unanticipated
problem since it is unexpected, related to the research, and represents
a more serious risk of harm to subjects than was previously known or recognized.
Occasionally
an incident, experience, or outcome occurs in research that does NOT meet
this definition of an adverse event. Consider, for example, if Charlie,
a study coordinator, leaves his laptop containing subjects’ identities
and social security numbers on the MBTA. This event would not meet the
definition of an adverse event, but would be a reportable unanticipated
problem since it is unexpected, related to the research, and represents
a more serious risk of harm to subjects than was previously known or recognized.
The following Venn diagram, from the OHRP guidance, summarizes the general relationship between AEs and unanticipated problems. Most AEs are not unanticipated problems. And, some unanticipated problems are not AEs. Again, only events that meet all 3 criteria of unanticipated problems must be individually reported to the IRB.
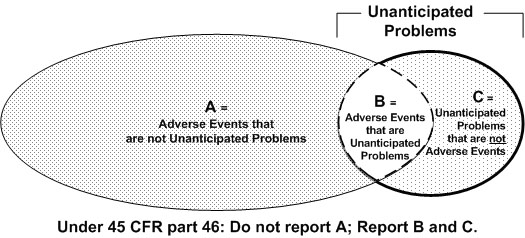
Reporting Unanticipated Problems to the IRB
Effective immediately, BUMC investigators will no longer individually report all SAEs to the IRB. Instead, only those events that meet the definition (3 criteria) of unanticipated problems will be individually reported to the IRB. These unanticipated problems, whether they are internal or external, must be reported to the IRB within two business days of the investigators learning of the event.
Internal events are those experienced by subjects enrolled by BUMC investigators no matter where the “event” occurs. For single site studies, all events are internal events. Only those internal events that meet the definition of unanticipated problems need to be individually reported to the BUMC IRB.
 External
events are those that occur to subjects enrolled at other (non-BUMC) sites.
Investigators in multi-center trials receive SAE reports of all external
SAEs from all sites. These external SAEs no longer have to be routinely
reported to the BUMC IRB on individual SAE reports. Instead, external
SAEs will ideally be reviewed by a monitoring entity (e.g., the research
sponsor, a coordinating or statistical center, a DSMB/DMC, an AE monitoring
committee) or the investigators, in accordance with a monitoring plan
described in the IRB-approved protocol. These entities should make the
determination as to whether the SAE represents an unanticipated problem.
Instances when external SAEs are determined to be unanticipated problems
should be rare; but when they occur, they must be submitted to the IRB.
The submission must include an explanation from the investigator or sponsor
as to why the event is considered an unanticipated problem, and
a description of the proposed protocol changes or other corrective actions
to be taken. [Note: Problems that are unexpected, related or possibly
related and represent a greater risk to subjects will almost always require
modification to the protocol and consent].
External
events are those that occur to subjects enrolled at other (non-BUMC) sites.
Investigators in multi-center trials receive SAE reports of all external
SAEs from all sites. These external SAEs no longer have to be routinely
reported to the BUMC IRB on individual SAE reports. Instead, external
SAEs will ideally be reviewed by a monitoring entity (e.g., the research
sponsor, a coordinating or statistical center, a DSMB/DMC, an AE monitoring
committee) or the investigators, in accordance with a monitoring plan
described in the IRB-approved protocol. These entities should make the
determination as to whether the SAE represents an unanticipated problem.
Instances when external SAEs are determined to be unanticipated problems
should be rare; but when they occur, they must be submitted to the IRB.
The submission must include an explanation from the investigator or sponsor
as to why the event is considered an unanticipated problem, and
a description of the proposed protocol changes or other corrective actions
to be taken. [Note: Problems that are unexpected, related or possibly
related and represent a greater risk to subjects will almost always require
modification to the protocol and consent].
It is important to note that the purpose of this article is to discuss
the reporting of events to the IRB. Investigators may have additional
responsibilities for reporting to the sponsor, the FDA, the Data Safety
Monitoring Board (DSMB), the Data Monitoring Committee (DMC), the Data
Coordinating Center (DCC), the Adverse Event Monitor, etc. The changes
to the IRB reporting policy discussed in this article do not change those
requirements. Note, the November 2007 issue of the CR Times will feature
an article about these additional reporting requirements.
Reporting Unanticipated Problems in INSPIR
 The
INSPIR SAE report will soon be renamed the “Unanticipated
Problems and Summary of Events Report”
(UPSER). The UPSER will be used to report all events that meet the definition
of unanticipated problems to the IRB (within two business days
of the investigator learning of the incident).
The
INSPIR SAE report will soon be renamed the “Unanticipated
Problems and Summary of Events Report”
(UPSER). The UPSER will be used to report all events that meet the definition
of unanticipated problems to the IRB (within two business days
of the investigator learning of the incident).
The UPSER may be submitted by any member of the study staff at any time, even when an amendment or progress report is pending.
Submitting Safety Monitors' Reports
Investigators will also use the UPSER to submit reports from safety monitoring entities (DSMBs, DMCs, AE Monitors, DCCs, etc.). These entities meet on a regular and as-needed basis that is pre-defined in the Data Safety Monitoring Plan. The reports typically document when the meeting took place, what was reviewed, and whether any changes to the protocol or consent are needed based on review of all AEs. Safety monitors’ reports should be submitted to the IRB as soon as the investigator receives them. It is very important that the most recent safety report is submitted prior to or at the same time as the Progress Report. If the safety monitor’s report is not available for the continuing review of the protocol, the approval of the study for continuation may be delayed.
Progress Reports and Summary Reports
For single center and multi-center studies where there is no DSMB or other formal safety monitoring body, the investigator must submit an AE summary report to the IRB. The AE summary report must include all AEs (including SAEs) and unanticipated problems that have occurred and must be submitted prior to or at the same time as the progress report. For studies with very few AEs and unanticipated problems, these can be summarized and evaluated by the PI in Section PR4 of the Progress Report. For studies with numerous AEs and unanticipated problems, investigators must submit a separate AE summary report using the UPSER form. (AE summary reports should not be attached to the protocol in Section S.)
The format of the AE summary reports may vary. Some may list each occurrence
individually while others may summarize AEs according to categories (i.e.,
5 headaches, 3 episodes of nausea, 2 leg pain, etc.) Each summary report
MUST include an evaluation by the PI (or the person designated in the
protocol as the AE monitor) as  to
whether the events, in total, suggest that the research places subjects
or others at a greater risk of harm (including physical, psychological,
economic, or social harm) than was previously known or recognized because
of their nature or their frequency of occurrence.
to
whether the events, in total, suggest that the research places subjects
or others at a greater risk of harm (including physical, psychological,
economic, or social harm) than was previously known or recognized because
of their nature or their frequency of occurrence.
For single center and multi-center studies where there is a DSMB, DMC, or other formal safety monitoring body approved by the IRB to be the safety monitors, the PI may provide a formal report from that monitoring body in lieu of submitting a summary of all AEs to the IRB.
IRB Reporting to OHRP and the FDA
OHRP and FDA regulations require that IRBs notify institutional officials, sponsors, OHRP, and the FDA of all internal unanticipated problems. Unlike reports of serious or continuing non-compliance (CR Times – April 2006), these reports do not indicate any deviation or wrong doing on the part of the investigators; but, instead, are made to inform federal agencies and sponsors of potential changes in risk in previously-approved research due to unanticipated problems. The IRB provides copies of these reports to the PI.
The BUMC IRB has significantly modified its policy for reporting SAEs to the IRB based on new regulatory guidance from OHRP and the FDA.
Additional Materials
- Reporting Algorithm
- Reporting Table
- Reporting Cheat Sheet
- New Educational Materials posted on IRB Website
Quiz
This Quiz applies to the recertification period from July 1, 2007 to June 30, 2009. CME credits are no longer offered or available as of 9/15/2010.
Click
here and close window if you are a BUMC researcher
and would like to take the quiz now.