![]()
![]()
Feature Article
Research Repositories (Part II) - IRB Review of Repositories
May 2013 Issue
By Mary A. Banks, BS, BSN,
Director, BUMC IRB
Authors has nothing to disclose with regards
to commercial support.
- Introduction
- Repository Infrastructure
- IRB Review
- Risks to subjects
- Informed consent and Data and Safety Monitoring
- Confidentiality and Additional Protections
- Summary
- Quiz
 This month’s feature article is part of a series dedicated to the topic of research repositories. The April 2013 article provided an overview of repositories and a discussion of the requirements for “recipient investigators” wishing to obtain data and/or samples from an existing repository. This feature article will focus on the IRB considerations when reviewing research that involves establishing a research repository. Since this article is Part Two of the series, if you haven’t already done so, it is recommended that you first read the April 2013 issue before proceeding with this article.
This month’s feature article is part of a series dedicated to the topic of research repositories. The April 2013 article provided an overview of repositories and a discussion of the requirements for “recipient investigators” wishing to obtain data and/or samples from an existing repository. This feature article will focus on the IRB considerations when reviewing research that involves establishing a research repository. Since this article is Part Two of the series, if you haven’t already done so, it is recommended that you first read the April 2013 issue before proceeding with this article.
Any investigator intending to establish a research repository must have in place the appropriate infrastructure to ensure that operations will run smoothly, that there is regulatory compliance, and that there are sufficient resources to ensure the confidentiality and the quality of the data/samples.
The following are some of the components of a repository infrastructure:
- An appropriate system for tracking intake and release of data/samples
- A stewardship mechanism that ensures that the repository is managed by qualified investigators
- A set of policies and procedures that address the operations of the repository
- A plan for monitoring and oversight of the repository, including quality monitoring
- A security plan for protection of the data
- A backup plan for storage of the samples (e.g., if there is power loss to the freezer)
- Available forms/agreements such as Materials Transfer Agreements, Data Use Agreements, and an understanding of when use of these is necessary
- An appropriate budget to cover the costs of operation
Since IRB approval is necessary for research repositories, the IRB reviews the aspects of the repository that impact human subjects’ protections. For example, the IRB will consider the plan for storage of subjects’ data because if the plan for data storage is insufficient, then subjects who participate in the repository research are at additional risk. The rest of the article will specifically address factors that the IRB considers in its review of research repositories.
The Banking Analogy
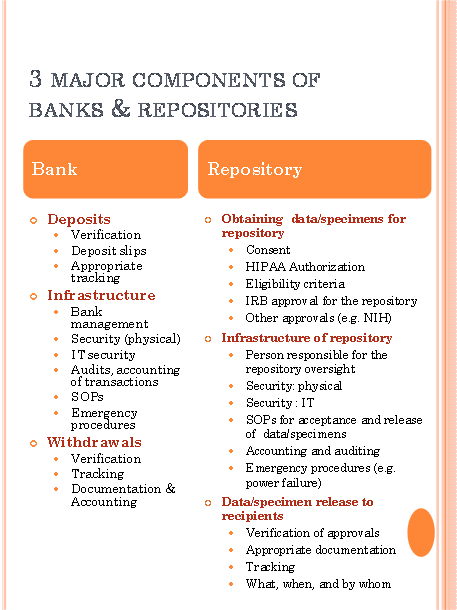
In order for the IRB to approve research (including research repositories), the IRB must determine that all of the requirements under Health and Human Services Regulations 45CFR46.111 (and, if applicable, the FDA regulations 21CFR 56.111) have been met. IRB review of research repositories is done either by expedited review or by the convened (full board) IRB. In most cases, especially when the research involves genetic research, the repository protocol is initially reviewed by the convened IRB. Subsequent reviews may be expedited depending on the determinations of the full board. At BUMC, repository protocols are submitted for IRB review via INSPIR. Click here to link to specific INSPIR instructions for repository research.
Risks to subjects are minimized
The IRB must consider all reasonably foreseeable risks related to the repository, not just the risks of physical harm. The IRB looks at which data/specimens are being obtained and considers:
- What are the physical risks?
- Are all risks described related to the research sample collection, including the risks of taking additional samples (e.g., extra tissue beyond clinical biopsy, additional collections from bronchoscopy or tissue biopsy for research, etc.)?
- Is it clear which are the research risks vs. Risks related to the clinical procedure?
- Are there non-physical risks?
- Risk of criminal or civil liability (i.e., legal trouble)
- Risks of damage to financial standing
- Risks of impact on employability , insurability, or reputation
Does the repository involve “sensitive” data/information/specimens?
- Is there a plan to collect/retain data about:
- Use of illegal drugs, underage drinking or alcohol abuse
- Child or elder abuse, sexual behavior
- “Sensitive” diseases, conditions, treatments related to HIV, psychiatric conditions, sexually transmitted diseases
- Genetic testing and test results
- Does the sensitivity of the information reasonably place repository “donors” at risk of non-physical harms?
- Genetic information: Repositories that involve genetic samples/genetic information or samples for future genetic testing are potentially greater risk due to:
- Incidental findings
- Social family connections/associations
- Potential impact on insurability and employability
- Potential future re-identification, even if the sample does not contain identifiers
Additionally, the IRB must determine whether the risks of repository participation are minimized. Therefore, all potential risks related to the research repository should be clearly identified in the research protocol, including risks for “additional” sample/data collection beyond clinical care. The IRB application must then specify how each of the risks will be minimized; for example:
- By having requests by recipient investigators for data/samples also be reviewed by a data use committee
- By having strict SOPs for ensuring that data/samples will only be released to recipients for use consistent with intent of subjects
- By ensuring that access to samples is controlled by well-designed repositories
- By tightly controlling access to identifiable information--especially when data/specimens include sensitive information
Risks to subjects are reasonable in relation to anticipated benefits
In most instances, repositories are not expected to have direct benefit to subjects. The protocol must, however, include an explanation as to any potential benefit to generalizable knowledge will be attained from the repository. Some additional points related to risk/benefit:
- Benefits shouldn’t be overstated.
- If collection of repository specimens involves greater than minimal risk, then the potential benefit must be proportional to the risk (e.g., if a biopsy is being done, then there should be planned analysis rather than storage for “maybe” future use).
- Potential benefit to “science” has implications for repository infrastructure. There must be sufficient protections for the specimens to help ensure that they will have some future research value. If samples are improperly stored or destroyed and not usable, then the risks outweigh the benefits because there are no benefits to subjects or science.
Selection of subjects is equitable
 The INSPIR protocol must clearly specify the eligibility (inclusion and exclusion) criteria for the repository. There must be a plan in place to ensure that only eligible subjects’ data/samples are “deposited” in the repository. The repository protocol should address the following points:
The INSPIR protocol must clearly specify the eligibility (inclusion and exclusion) criteria for the repository. There must be a plan in place to ensure that only eligible subjects’ data/samples are “deposited” in the repository. The repository protocol should address the following points:
- What are the eligibility criteria for repository participation?
- How will potential subjects (donors) be identified and recruited?
- If samples/data will not be obtained directly from subjects, then where are data/specimens coming from?
- Will there be screening of potential donors?
- Will subjects be approached by their clinicians or by the researchers?
- Will non-English speaking subjects be recruited? If not, why not?
Informed consent will be sought from each prospective subject or the subject’s legally authorized representative
It is important to note that “research informed consent” by regulatory definition means a research informed consent that has been IRB-approved and contains all the required elements of consent, unless some elements have been waived by the IRB. A general statement in a clinical/surgical consent that says that “we can keep your samples for research” does NOT qualify as research informed consent.
Obtaining informed consent from each individual subject for participation in a repository is the “gold standard”. The IRB recognizes that sometimes this is not possible, especially in situations such as:
- Previously-collected samples from non-research situations such as clinical care
- Previously-collected samples from other research
- Research previously approved where future use of data/specimens was not considered at the time of consent
In these situations, the IRB will consider whether or not it is appropriate to waive the requirement for informed consent for placement of the samples/data into a repository. In order to do so, the IRB must be able to determine that the repository research meets the regulatory criteria for waiver of informed consent. This is frequently an issue for studies involving retrospective samples/data where the repository uses were not considered at the time that the data/samples were obtained. The factors that the IRB will consider when making a waiver determination are what the potential subjects expected when they initially provided their data/specimens, the perceived risks (does the repository involve sensitive data, genetic material, etc.), and the planned uses (e.g., expansive release outside the institution, commercial uses, etc.).
Each repository protocol should address:
- Who will obtain consent from subjects and under what circumstances?
- Obtaining informed consent for research is a research activity and should not be delegated to non-researchers (e.g., clinical staff)
- Who will provide consent: The subject only or will consent be sought from the legally authorized representative for decisionally impaired subjects?
- Subjects must be given sufficient time to consider participation.
- How will the repository be set up to ensure that:
- Data/specimens are only collected/retained from those who gave consent?
- When the consent has multiple Opt-in/Opt-out options, how will the repository be managed to ensure these selections are honored?
The informed consent document for repository research will contain the “standard” elements that are typically seen in research consent forms such as background, purpose, what happens, risks, benefits, alternatives, confidentiality, payment information and subjects rights. Because of the nature of repository protocols, there are additional elements that may also be required, depending on the repository, such as:
- General description of a repository and genetics
- Purpose of this repository: Why are subjects being asked to participate
- Potential uses (opt-in /opt-out options) by:
- Other BUMC investigators
- Researchers at other academic institutions
- National repositories; e.g., NIH dbGaP, NCI
- Commercial entities/industry
- GINA language
- Certificate of Confidentiality (CoC) language
- Research results should not be expected to be returned to the subjects
- What would happen if there are “incidental findings”
- Commercial use language
Informed consent will be appropriately documented
 As a rule, documentation of consent (subjects’ signatures) is required for repository consent forms. As discussed above, the IRB will determine on a case-by-case basis whether it is appropriate to allow for consent by LAR for participation in the repository.
As a rule, documentation of consent (subjects’ signatures) is required for repository consent forms. As discussed above, the IRB will determine on a case-by-case basis whether it is appropriate to allow for consent by LAR for participation in the repository.
Data and Safety Monitoring
The extent of the proposed Data and Safety Monitoring Plan (DSMP) will depend on the risks of the repository protocol. As a rule, repositories do not usually require a Data and Safety Monitoring Board (DSMB); however, many larger repositories (for example, the Framingham Heart Study) have data use committees. All repositories should have a DSMP. Minimally, the DSMP should address:
- Procedures for overseeing and monitoring data/specimens
- Security procedures
- Emergency protections for samples/specimens (e.g., power loss)
- Delegation of responsibilities for oversight, monitoring, reporting unanticipated problems to the IRB, etc.
Most repositories collect and store some data. It is important that the IRB protocol clearly describe all the data points that will be part of the repository. It should also be clear which identifiers (if any) will be associated with the data/samples, and whether the data/samples will be coded; and if so, who will have access to the master code to link the data/samples with the identifiers. Most repository protocols run into problems during IRB review because the confidentiality section is not clearly described. The repository protocol should specifically detail each of the following:
- Which identifiers will be “collected” with the data/specimens
- Which identifiers will be stored with the data/specimens, and who specifically will have access to these
- Which identifiers (if any) will be released to recipient investigators
Repository investigators should carefully read the BUMC Privacy and Confidentiality module and ensure that appropriate terminology is used when describing confidentiality protections in their INSPIR protocol. This module discusses in detail the various HIPAA requirements, which also apply to repository research.
In some instances, the IRB will require that the investigator obtain a Certificate of Confidentiality to add additional protection for the repository. In repository studies involving genetic research, the IRB will frequently require that Genetic Information Non-Discrimination Act (GINA) language be added to the consent form.
Additional Protections for Vulnerable Subjects
The repository protocol must identify potentially vulnerable subjects who might be asked to participate in the repository. The IRB will then make determinations as to what additional safeguards are necessary (for example, the consent of both parents for higher risk studies) to protect the vulnerable subjects. When the repository involves collection of data/samples from children, the determination must be made as to whether these subjects will be contacted for reconsent when they reach the age of majority. It is important to note that genetic research and repositories often involve difficult concepts, so the investigators must be clear about how they are going to help the subjects understand the risks.
This article outlines the regulatory criteria for IRB approval of research, as well as questions and concerns that the IRB considers when reviewing repository research.
Quiz
There are no quiz questions this month. Take a few minutes and check where you stand with quizzes that count towards Recertification. Click here for an explanation of who needs to be recertified and how to meet the requirements. If you still have questions, please send them to crtimes@bu.edu.