Director, Office of the Institutional Review Board

By Mary A. Banks, RN, BS, BSN
Director, Office of the Institutional Review Board
Introduction
Reporting Requirements Vary
Classification of Adverse Events
Reporting Aes/SAEs to the IRB
Summary
Introduction
This morning one of your study subjects has come to the office for a blood draw. He happens to mention that he recently spent a couple of days in another hospital. Should you report this hospitalization to the IRB? If so, when should you report it, immediately or at some later point?
If you are confused by the conflicting requirements surrounding adverse event reporting, you are not alone. This is a problem that has been identified by investigators nationwide. You may be surprised to learn that this problem is finally getting some attention on a Federal level. The FDA has announced that there will be a hearing in March, 2005 to address the complexities surrounding adverse event reporting (link to announcement) . In the meantime, here at BUMC the IRB Executive Committee has recently decided to modify the local adverse event reporting requirements for investigators. The goal is to simplify the reporting process for BUMC investigators and decrease the administrative burden for investigators and the IRB so that both groups can focus on the more severe, related events that are most likely to impact the risk level of studies.
Depending on where you look, you will find definitions of reportable adverse events (Aes) and serious adverse events (SAEs) that vary significantly.
The FDA describes an unexpected adverse drug experience to be "any adverse drug experience, the specificity or severity of which is not consistent with the current investigator brochure or….. The general investigational plan". FDA regulations (21 CFR 312 ) require that sponsors notify the FDA and participating investigators of any adverse event associated with a test article that is “both serious and unexpected”.
The Office of Human Research Protection (OHRP) in 45 CFR 46 Subpart A requires that each IRB has written procedures and policies for ensuring reporting of “unanticipated problems” involving risks to participants.
Previously, BUMC policies have required investigators to report ALL adverse events that occurred to subjects during research. With the new policy the IRB will be asking investigators to make certain determinations regarding each adverse event. Based on these determinations the investigator may or may not be required to report the event to the IRB or may only need to report the event at the time of the progress report.
This change places more responsibility on the shoulders of the investigator. In the eyes of the IRB, it is the investigator who is in the best position to make these decisions because the investigator is most knowledgeable about the study itself, the underlying disease and the condition of the individual subjects.
Classification of Adverse Events
In order to easily follow the new guidelines for reporting adverse events investigators must understand how the IRB categorizes these events. Described here are the various ways that the IRB evaluates adverse events and the way the IRB wants events to be reported.
Severity
The first determination the IRB asks the investigator to make is whether an event is Serious or Non-serious. The investigator must grade the event from Grade 1 to Grade 5 depending on its severity while giving consideration to the individual circumstances surrounding the event. For example, a subject has been admitted to the hospital with chest pain. In some cases this event might be determined to be “severe” and in other instances the event might be determined to be “life-threatening.”
Non-serious (Aes) Grade 1= mild Grade 2= moderate |
Serious (SAEs) Grade 3= severe Grade 4= life-threatening Grade 5= fatal |
Ownership of the AE/ SAE
Aes and SAEs are categorized as either Internal or External. This determination is made based on whether the subject “belongs” to the local PI or whether the subject is a subject in a multi-center study “under” a different PI. As a general rule, if the study was approved by the BUMC IRB and the subject signed a consent form approved by our IRB, then the subject “belongs” to BUMC.
A note of caution, the determination as to whether an adverse event is internal or external should NOT be made based on where the actual event occurred. For example, if a subject is enrolled in a cardiac study under a local BUMC investigator but is then admitted to a hospital with chest pain while on vacation in California , this event must be treated as an Internal SAE and should be reported as such to the BUMC IRB.
Relatedness
The investigator must also decide if the AE/SAE is “related” to the research. The IRB is interested in whether, in the determination of the investigator, the event is related to ANY aspect of the research protocol (i.e. any of the research procedures or interventions).
The IRB asks the investigator to make this determination because, by knowing if the event is related to the research rather than to the subject’s underlying condition, the IRB can better evaluate whether the risks of the study have changed and whether the study must be changed or terminated or whether the consent form requires modification. The IRB does not want to be in the position of suspending research or requiring that risks be added to consent forms because of events that have occurred that are not related to the research. This could cause subjects to become confused and might unnecessarily discourage them from participating in the research. For example, consider a study looking at cardiac disease in people with cardiac risk factors where the only two research interventions are drawing blood and measurement of blood pressure. If two days after a study visit a subject is admitted to the hospital with chest pain the investigator would most likely conclude that the event is unrelated to the research (since it is unlikely that the chest pain was caused by the blood drawing or the blood pressure measurements).
As part of the initial protocol submission to the IRB investigators must list all of the research related procedures or interventions of the study in Section F2 of the INSPIR application. Investigators should reference this section of the IRB protocol when making their determination as to whether the event in question is Related or Unrelated to the research. Relatedness is subdivided into 5 categories.
Unrelated Unlikely related Not related |
Related Probably Possibly Likely |
Expectedness
Finally, the investigator must decide if the AE/ SAE is an “expected” outcome of the research. Some events happen to people on research protocols that may be anticipated. An obvious example of this would be a subject who experiences nausea after receiving a study drug that is known to cause nausea. Certainly in this case the subject should have been informed of the risk of nausea in the informed consent and nausea should have been listed in the IRB protocol as a risk of participation in the research. This would be considered an Expected event.
The IRB has determined that, if an event is listed as a risk in the IRB protocol (Section H of INSPIR) and in the “Risks” section of the consent form, then that event is considered “Expected.” Events that are not listed in those locations are considered to be “Unexpected.”
It is however important to note that some events must be reported as Unexpected because they occur with more frequency, in greater duration or with greater severity than what has been anticipated. For example, in the situation described above, the protocol and consent form state that the subject can anticipate mild to moderate nausea for 3-5 days following the infusion of the study drug. If this subject is admitted to the hospital 7 days following his infusion for severe vomiting and dehydration requiring IV hydration then this event should be categorized as Unexpected as the severity and duration of the nausea exceeded what was described in the risk section of the protocol and consent form.
Reporting Aes/ SAEs to the IRB
Once the PI has made a determination as to how to categorize an event; Serious or Non-serious, Internal or External, Related or Unrelated and Expected or Unexpected then it should be relatively easy to use the following diagrams to determine when and if the event needs to be reported to the IRB.
This is a brief summary of the changes that are reflected in these diagrams. For complete details of the changes click here.
Diagram #1 demonstrates the reporting process for External Adverse events and DSMB reports (whether internal or external). Diagram #2 demonstrates the reporting process for Internal Adverse Events.
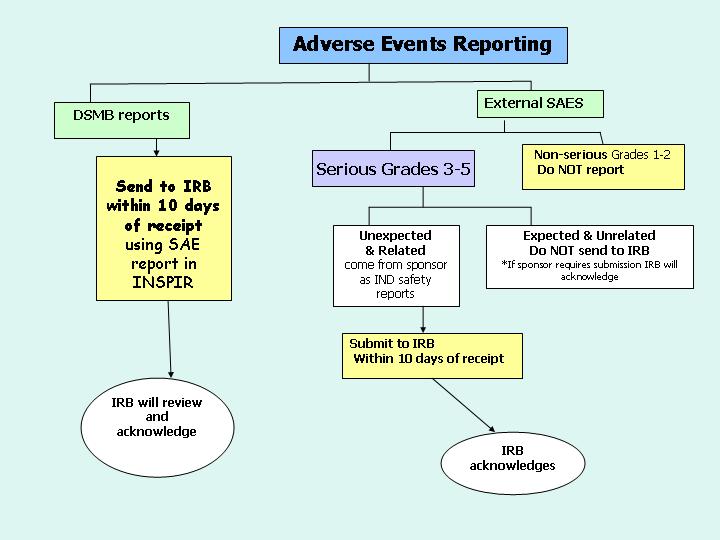
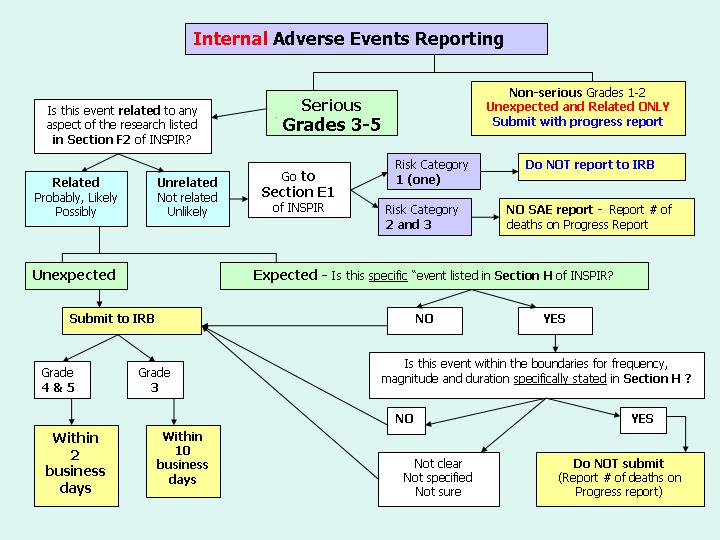
These reporting requirements apply to all IRB studies, those that are minimal risk (Expedited and Exempt), as well as those that are greater than minimal risk (Full Board Studies). Reporting adverse events to the IRB does not in any way change the PI’s responsibilities for other reporting of such events to others such as the FDA or the study sponsor. More details regarding the specifics of adverse event reporting can be found on the IRB website under SAE reporting.
It is the responsibility of the IRB to review the risks of research protocols on an ongoing basis to determine whether the risk benefit ratio of a study has changed and whether there has been a change in risk that could impact a subject’s willingness to participate in the research. When an adverse event occurs the IRB relies on the investigator to appropriately evaluate and report the event so that this decision can be made. The IRB has developed a modified reporting system to facilitate this process.